
























快速掌握欧盟医疗器械新法规MDR重点,你了解多少?




欧盟医疗器械法规(MDR) [REGULATION (EU) 2017/745](以下简称”MDR”)于2017年5月25日生效,并将于2020年5月26日取代医疗器械指令(MDD,93/42 / EEC)和有缘可植入医疗器械指令(AIMDD,90/385 / EEC)。
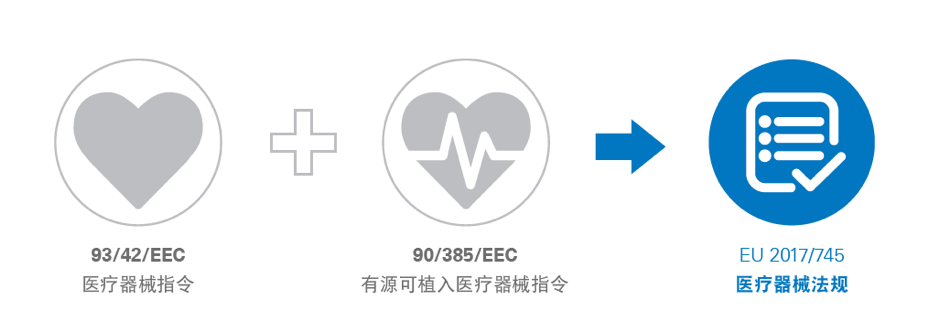
MDR的法规过渡期为三年。这意味着所有新器械的制造商及其利益相关者必须在2020年5月26日期强制符合MDR的要求,制造商应在过渡期内更新技术文件和流程以满足法规要求。之前获得MDD/AIMD证书的CE认证产品,最晚在2020年5月26日后,就无法在欧盟市场贩卖或安装。我们简单地梳理了MDD到MDR过渡期的时间轴如下:
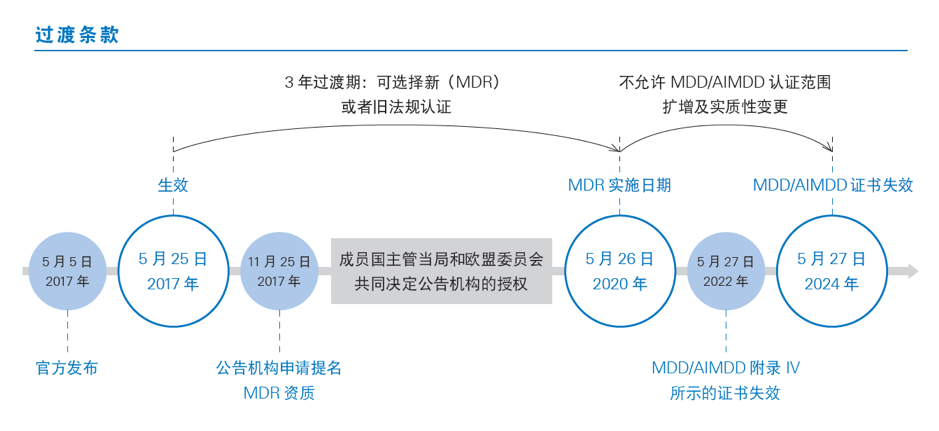
依据Article 120 clause2 的规定,过渡期内公告机构签发的CE证书继续有效,但是从其交付日期起有效期不超过5年,并且于2024年5月27日失效。
主要变更
MDR新规的法律框架较完整,各项规定比之前的指令更全面详细,对相关企业及从业人员提出更严格的要求。其主要变更如下:
1. 更广泛的应用领域(包含非医疗区域)
2. UDI:每台医疗设备的唯一之识别码
3. 更严格的技术文挡(TD)要求
4. 更严格的负责人要求:医疗器械的专业知试
5. 高风险医疗机械的新审查程序
6. 更严格的临床评估和测试要求: 临床数据的收集持续到产品上市后
7. 更严格的公告机构监督: 新公告机构的选择与检查
8. 欧洲医疗器械数据库: 更加透明且便于信息流通的数据库
此外,公告机构应至少每5年对证书持有者(医疗器械制造商)进行一次飞行检查,目的在于保证医疗器械产品质量的确定性和稳定性,让制造商始终在遵守MDR法规的情况下生产医疗器械。
TUV莱茵专家建议制造商

TÜV莱茵,您值得信赖的伙伴
作为全球领先的检测,检验和认证服务机构,我们在140多年时间里一直致力于为客户提供产品支持服务。2019年9月26日TÜV莱茵正式成为MDR的公告机构,可为出口欧盟的医疗器械产品提供新法规的符合性评估服务(CE认证)。作为欧洲最大的公告机构之一,我们代表着稳定、可靠和最高质量的服务水准。我们的全球专家网络在体外诊断医疗器械/医疗器械和质量管理体系认证方面拥有广博的知识和丰富的经验,可为客户提供一站式的认证服务。如有更多关于医疗器械测试、认证等相关问题,请与我们联系。


推荐文章
关注我们
4001183833 / 8009993668
Service-gc@tuv.com
www.tuv.com
德国莱茵TÜV是一家国际领先的技术服务供应商。在全球近60个国家和地区设有500多个分支机构,拥有超过20,000名员工,服务涵盖工业服务与信息安全,交通服务,产品服务,管理体系服务和莱茵学院与生命关怀。自1872年成立以来,我们一直为解决人类、环境和科技互动过程中出现的挑战,提供安全的、可持续的解决方案。作为一个独立、公正和专业的机构,我们长期致力于营造一个同时符合人类和环境需要的美好未来。
热门分享

