
























新版医疗器械管理体系标准ISO 13485应对指南


朱丹

TÜV莱茵大中华区医疗器械服务部项目经理 | 在医疗器械研发、质量管理和认证领域拥有15年以上的丰富经历,作为审核员多次代表TÜV莱茵接受并通过中国、德国、加拿大、日本、巴西等国监管部门的目击审核。
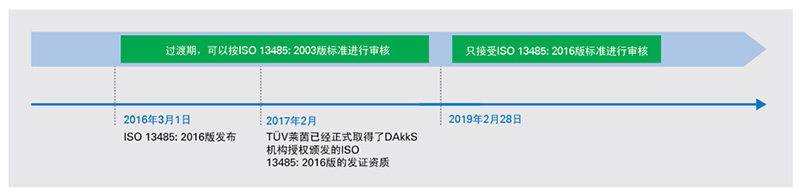
医疗器械管理体系新版标准ISO 13485:2016已于2016年3月1日正式发布,新旧标准的过渡期将持续到2019年2月28日。为了让更多企业及时了解新版本的关键点,TÜV莱茵为大家详述各关键时间点、新旧版本的显著变化,并提供应对指南,为企业顺利转版支招。
新版本的显著变化一览
1、 导入生命周期的概念。
2、 更加明确了标准的适用范围,增加了适用于医疗器械全生命周期各阶段(研发、生产、上市后活动)的医疗器械组织。
3、 增加了适用于供方或其他服务供方等要求。
4、 增加到了20个定义,并细化了一些现有定义。如:授权代表、临床评价、抱怨、经销商、进口商、生命周期、制造商、医疗器械族、性能评价、上市后监督、采购产品、风险、风险管理、无菌屏障系统等专业术语。
5、 增加了“法规要求”术语的次数,一旦使用术语“法规要求”时,其涵盖了适用于本标准用户的任何法律法规的要求。
6、 强化了企业满足法规要求的主体责任。
7、 加强了风险管理,不仅对医疗器械产品和服务的全生命周期实施风险管理,而且明确了对质量管理体系的过程实施风险管理的要求,提出“应用基于风险的方法控制质量管理体系所需的适当过程”。
8、 新增了“组织应将用于质量管理体系的计算机软件应用进行确认的要求”。
9、 提出在供方评审时应基于风险的考量
10、 新标准要求应当和供方签订书面质量协议,适用时,应当与供方(原料和服务)签订书面质量协议,规定变更告知的义务。
11、 强调了如果组织发现采购的产品有任何变化,需要评估此变化所带来的影响。
12、 提出从反馈信息中识别改进机会,输入到风险管理以及纠正预防措施中。
13、 提出反馈信息应包括来自生产阶段的内部反馈。
14、 新标准强调了抱怨处理的重要性。
15、 新增“8.2.3向监管机构报告”条款
16、 新标准中“形成文件”达到56处,保持记录要求48处。新增有关文件要求的条款,如4.2.3医疗器械文档;7.3.10设计和开发文档。
企业如何应对?TÜV莱茵为你提供行动方案
1. 获取一份标准正式版。
2. 新老版本差异比对,并进行“差距分析”文档建立。如在审核中发现缺少“差距分析”,将会被记为重大不符合项。
3. 确认并分配体系变更所需要的资源,包括人力资源 。
4. 由于新老标准差异涉及到多个部门,TÜV莱茵建议增加内审次数,以确保能符合ISO 13485:2016的要求。
5. 了解认证和过渡期的时间表。TÜV莱茵建议您尽快与我们联系进行证书升级。新客户或者是换证客户我们将采用ISO 13485:2016标准来进行审核。
6. 制定“质量计划”,列明需要完成的工作明细 。“质量计划”也是认证审核中需要检查的文档。
7. 为相关人员提供新标培训,沟通“质量计划”。如果您需要第三方机构为您提供预审,TÜV莱茵将随时为您服务。
8. 确保您的内审涵盖到“差距分析”中所列明的所有变更部分。
9. 准备接受公告机构的升级审核。
TÜV莱茵已于2017年2月被DAkkS认可,拥有ISO 13485:2016版的发证资质。



关注我们
4001183833 / 8009993668
Service-gc@tuv.com
www.tuv.com
德国莱茵TÜV是一家国际领先的技术服务供应商。在全球近60个国家和地区设有500多个分支机构,拥有超过20,000名员工,服务涵盖工业服务与信息安全,交通服务,产品服务,管理体系服务和莱茵学院与生命关怀。自1872年成立以来,我们一直为解决人类、环境和科技互动过程中出现的挑战,提供安全的、可持续的解决方案。作为一个独立、公正和专业的机构,我们长期致力于营造一个同时符合人类和环境需要的美好未来。
热门分享

